How to use#
Installation#
The full name of pattools is pattools-methy. This naming was chosen because the name pattools was already taken on PyPI. There are several ways to install pattools
Install from PyPI#
pip install pattools-methy
Install from github#
pip install git+https://github.com/hcyvan/pattools.git
Install from source#
Download the source code, and then
cd /path/to/source/code
pip install -r requirements.txt
python ./setup.py install
Usage#
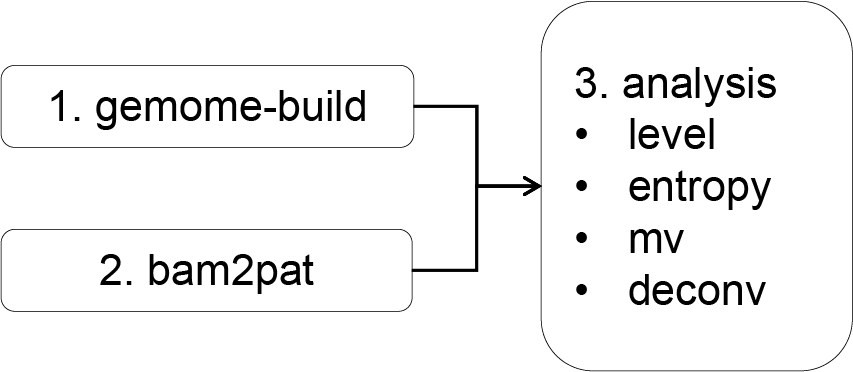
Once the installation is complete, you can begin analyzing BS-seq data. The general workflow involves the following steps:
Prepare the Reference Genome: Build the appropriate reference genome based on the species being analyzed.
eg:
pattools reference -f /PathTo/hg38.fa.gz -o /OutputPathTo/hg38 -c chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19,chr20,chr21,chr22,chrX,chrY,chrM
Convert Data to PAT Format: Convert the BS-seq data to the PAT format for compatibility with further processing.
Conduct Downstream Analysis: Perform additional analysis steps based on your specific research needs and objectives.